Manejo del Síndrome Antifosfolipídico
Se caracteriza por la aparición de trombosis vasculares y abortos recurrentes, relacionadas directamente con la presencia de anticuerpo antifosfolipídico .
Autor: Prof. Dr. José Manuel Porcel
Generalidades
Los anticuerpos antifosfolipídicos (AAF) son un grupo heterogéneo de autoanticuerpos que se unen a fosfolípidos (p.ej. cardiolipina) o a proteínas plasmáticas que tienen afinidad por superficies fosfolipídicas (p.ej. beta-2-glucoproteína I, protrombina, anexina). Los fosfolípidos están involucrados en la cascada de la coagulación sanguínea. La aparición de manifestaciones clínicas, fundamentalmente trombosis vasculares y abortos recurrentes, relacionadas directamente con la presencia de AAF define el síndrome antifosfolipídico (SAF) o síndrome de Hughes (en honor al reumatólogo inglés que lo describió en 1983).
En la mitad de los casos, el paciente con SAF no tiene evidencia de ninguna otra enfermedad autoinmune (SAF primario –SAFP-), mientras que en la otra mitad el SAF ocurre en asociación con otros trastornos autoinmunes, principalmente lupus eritematoso sistémico (LES) –el 30-50% de sujetos con LES tienen AAF- pero también artritis reumatoide, esclerodermia o enfermedad de Behçet (SAF secundario). Tanto en un caso como en otro, el autoantígeno contra el que van dirigidos los AAF suele ser la beta-2-glucoproteína I y no los fosfolípidos per se (AAF cofactor-dependientes). No obstante, los AAF pueden acompañar a multitud de otros procesos, como infecciones (virus de la inmunodeficiencia humana, hepatitis C, citomegalovirus), trastornos linfoproliferativos, uso de fármacos (clorpromazina, fenitoína, hidralacina, procainamida, tiazidas, anti-TNFα) o hemodiálisis, circunstancias en las que suele tratarse de anticuerpos IgM presentes en bajas concentraciones, dirigidos contra verdaderas estructuras fosfolipídicas (AAF cofactor-independientes) y que no se asocian con eventos trombóticos. También se han descrito AAF en el 5-10% de los donantes de sangre, aunque este porcentaje se reduce a menos del 2% si consideramos sólo a los sujetos con títulos persistentemente positivos.
En un estudio retrospectivo reciente de 128 pacientes con SAFP se observó que sólo el 14% desarrollaron un LES o una enfermedad similar al lupus después de un periodo de seguimiento medio de 8 años.
Detección de anticuerpos antifosfolípido en el laboratorio
En la práctica clínica los AAF que se detectan en todos los laboratorios son los anticuerpos anticardiolipina (ACL) y el anticoagulante lúpico (AL). Probablemente en el futuro inmediato se deba incluir la medición de los anticuerpos anti-beta-2-glucoproteína I.
Los ACL, bien sean del isotipo IgM o IgG, se detectan mediante una técnica de ELISA. Los resultados se expresan en unidades MPL y GPL, respectivamente, de una forma semicuantitativa como negativos (<10 GPL o MPL), positivos débiles (10-40 GPL o MPL), positivos medios (40-80 GPL o MPL) o positivos altos (>80 GPL o MPL). Sólo los valores a partir de positivo medio (>40 GPL o MPL) se incluyen actualmente en los criterios diagnósticos de SAF. Hay que señalar que todavía existe controversia y falta de consenso sobre qué valores de ACL se deben considerar como positivos a efectos diagnósticos. Cifras que oscilan entre 20 y 40 GPL o MPL obligan a descartar otras causas de trombofilia, antes de aceptarse como diagnósticas.
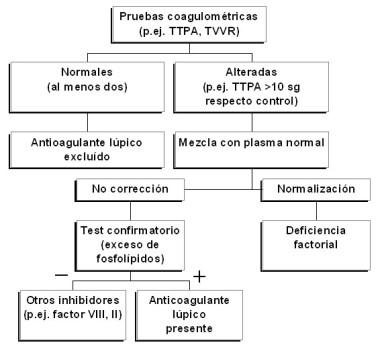
La detección de AL se efectúa mediante técnicas coagulométricas funcionales que ponen de manifiesto la presencia de anticuerpos dirigidos contra la fracción fosfolipídica del complejo activador de la protrombina. Para asegurar la presencia de AL se deben cumplir las siguientes 4 condiciones (Fig. 1):
Fig. 1: Detección del anticoagulante lúpico en el laboratorio
En la práctica clínica los AAF que se detectan en todos los laboratorios son los anticuerpos anticardiolipina (ACL) y el anticoagulante lúpico (AL). Probablemente en el futuro inmediato se deba incluir la medición de los anticuerpos anti-beta-2-glucoproteína I.
Los ACL, bien sean del isotipo IgM o IgG, se detectan mediante una técnica de ELISA. Los resultados se expresan en unidades MPL y GPL, respectivamente, de una forma semicuantitativa como negativos (<10 GPL o MPL), positivos débiles (10-40 GPL o MPL), positivos medios (40-80 GPL o MPL) o positivos altos (>80 GPL o MPL). Sólo los valores a partir de positivo medio (>40 GPL o MPL) se incluyen actualmente en los criterios diagnósticos de SAF. Hay que señalar que todavía existe controversia y falta de consenso sobre qué valores de ACL se deben considerar como positivos a efectos diagnósticos. Cifras que oscilan entre 20 y 40 GPL o MPL obligan a descartar otras causas de trombofilia, antes de aceptarse como diagnósticas.
La detección de AL se efectúa mediante técnicas coagulométricas funcionales que ponen de manifiesto la presencia de anticuerpos dirigidos contra la fracción fosfolipídica del complejo activador de la protrombina. Para asegurar la presencia de AL se deben cumplir las siguientes 4 condiciones (Fig. 1):
Fig. 1: Detección del anticoagulante lúpico en el laboratorio
1) Prolongación de al menos un tiempo de coagulación dependiente de fosfolípidos en el plasma del paciente objeto de diagnóstico. Para demostrar este extremo se pueden utilizar diversas pruebas, capaces de evaluar la vía intrínseca de la coagulación (tiempo de tromboplastina parcial activada -TTPA-, tiempo de tromboplastina parcial activada diluida, tiempo de coagulación con caolín), la vía extrínseca (tiempo de protrombina diluido), o la vía final común (tiempo del veneno de víbora de Russell diluido –TVVR-, tiempo de textarina y ecarina, tiempo de veneno de Taipan). La ausencia de una prolongación del TTPA (>10 sg respecto al control), el test más empleado, no excluye la existencia de un AL si se utiliza un reactivo poco sensible a los AAF. En tales circunstancias recurriremos a otra prueba de coagulación que evalúe una porción distinta de la cascada enzimática (p.ej. TVVR).
2) Fallo para corregir el tiempo de coagulación prolongado, después de mezclar el plasma del paciente con plasma normal (relación 1:1). Con ello se excluyen deficiencias de factores de la coagulación, contenidos en el plasma normal.
3) Acortamiento o corrección del tiempo de coagulación prolongado tras la adición de un exceso de fosfolípidos sintéticos o procedentes de un lisado plaquetario (prueba confirmatoria).
4) Exclusión de otras coagulopatías si el test confirmatorio es negativo (p.ej. inhibidores del factor VIII o del factor II). Paradójicamente, el AL prolonga los tiempos de coagulación in vitro, pero promueve la formación de coágulos in vivo.
Dado que en un 35% de los pacientes no coexisten los ACL y el AL (es decir, tienen un subtipo de anticuerpo u otro), es necesaria la realización de ambas pruebas para descartar fehacientemente la existencia de AAF. A diferencia de los ACL, la prueba de AL no es valorable en pacientes que estén sometidos a tratamiento anticoagulante; deberán haber transcurrido al menos 2 semanas desde la finalización de este último. Por otro lado, en algunos sujetos el AL también puede alterar ligeramente el tiempo de protrombina, lo cual repercutiría sobre el valor del INR (parámetro que se evalúa durante el tratamiento con dicumarínicos). También es posible que la existencia de anticuerpos anti-protrombina provoque un déficit funcional de esta proteína, circunstancia que generalmente se observa en asociación con AL. Cuando ello deriva en un sangrado significativo, a esta rara entidad la denominamos síndrome de AL-hipoprotrombinemia.
Dado que en un 35% de los pacientes no coexisten los ACL y el AL (es decir, tienen un subtipo de anticuerpo u otro), es necesaria la realización de ambas pruebas para descartar fehacientemente la existencia de AAF. A diferencia de los ACL, la prueba de AL no es valorable en pacientes que estén sometidos a tratamiento anticoagulante; deberán haber transcurrido al menos 2 semanas desde la finalización de este último. Por otro lado, en algunos sujetos el AL también puede alterar ligeramente el tiempo de protrombina, lo cual repercutiría sobre el valor del INR (parámetro que se evalúa durante el tratamiento con dicumarínicos). También es posible que la existencia de anticuerpos anti-protrombina provoque un déficit funcional de esta proteína, circunstancia que generalmente se observa en asociación con AL. Cuando ello deriva en un sangrado significativo, a esta rara entidad la denominamos síndrome de AL-hipoprotrombinemia.
Con menor frecuencia, en los pacientes con AAF se puede constatar la existencia de una serología luética falsamente positiva. Esto significa que la prueba reagínica es positiva, debido a que VDRL o RPR utilizan una mezcla de fosfolípidos como substrato, pero la prueba treponémica específica es negativa.
Manifestaciones clínicas
Los AAF, bien por su interacción con el endotelio vascular, con las plaquetas o por interferir con los complejos de proteínas y fosfolípidos que regulan la coagulación, promueven el desarrollo de trombosis. Este riesgo parece mayor en los sujetos que exhiben AL que en aquellos que sólo poseen ACL, especialmente si los títulos no son muy altos. Se ha estimado que el 50% de pacientes con LES y AAF desarrollan un SAF después de un periodo de observación de 20 años.
Manifestaciones clínicas
Los AAF, bien por su interacción con el endotelio vascular, con las plaquetas o por interferir con los complejos de proteínas y fosfolípidos que regulan la coagulación, promueven el desarrollo de trombosis. Este riesgo parece mayor en los sujetos que exhiben AL que en aquellos que sólo poseen ACL, especialmente si los títulos no son muy altos. Se ha estimado que el 50% de pacientes con LES y AAF desarrollan un SAF después de un periodo de observación de 20 años.
Trombosis vasculares
El SAF constituye uno de los estados de hipercoagulabilidad en los que puede producirse tanto trombosis venosas como arteriales. Los estudios poblaciones muestran que el SAF y la resistencia a la proteína C activada (factor V de Leiden) son las causas más comunes de trombofilia, representando cada una el 20% de los casos de trombosis recurrente en gente joven. El SAF y las trombofilias genéticas son proporcionalmente menos frecuentes en personas de más de 65 años, donde la edad, la arteriosclerosis y el cáncer son causas más habituales de trombosis.
El SAF constituye uno de los estados de hipercoagulabilidad en los que puede producirse tanto trombosis venosas como arteriales. Los estudios poblaciones muestran que el SAF y la resistencia a la proteína C activada (factor V de Leiden) son las causas más comunes de trombofilia, representando cada una el 20% de los casos de trombosis recurrente en gente joven. El SAF y las trombofilias genéticas son proporcionalmente menos frecuentes en personas de más de 65 años, donde la edad, la arteriosclerosis y el cáncer son causas más habituales de trombosis.
Las trombosis venosas profundas, especialmente de las extremidades inferiores, son la manifestación más común del SAF y, en la mitad de los casos, se acompañan de embolismos pulmonares. Las trombosis arteriales son menos frecuentes que las venosas; en cerca del 50% de casos afectan a la circulación cerebral (accidente isquémico transitorio o ictus), en el 25% a la coronaria (angina, infarto de miocardio) y en otro 25% a otros territorios como el cutáneo (úlceras, gangrena digital), retiniano o renal. Los episodios trombóticos relacionados con el SAF pueden tener lugar en lechos vasculares que se afectan infrecuentemente en otros estados protrombóticos y tienen tendencia a recurrir en el mismo sistema vascular (arterial o venoso) que el evento original.
Se debe tener en cuenta que no todos los episodios de isquemia arterial o infarto tienen un origen trombótico. Las embolias, procedentes de vegetaciones mitrales o aórticas, también son causa de ictus. De hecho, las anomalías valvulares cardiacas detectadas por ecocardiografía son muy frecuentes en pacientes con SAF (30-70%), aunque la mayoría de ellas tienen escasa trascendencia clínica. Suele tratarse de regurgitaciones que afectan a la válvula mitral o menos frecuentemente, aórtica. Las vegetaciones sobre dichas válvulas sólo se han descrito en el 4% de pacientes con SAF.
Los sujetos con SAF pueden desarrollar trombosis agudas o crónicas en la microcirculación (capilares, arteriolas, vénulas), que ocasionan una pérdida aguda o lentamente progresiva de la función de un órgano (p.ej. riñón). De hecho, en dos terceras partes de los pacientes con SAF asociado a LES se detecta nefropatía microtrombótica, que se manifiesta clínicamente como hipertensión arterial e insuficiencia renal. Una microangiopatía trombótica diseminada es la característica fundamental del llamado SAF “catastrófico”, como se describirá más adelante.
Morbilidad gestacional
Aproximadamente un 15% de las mujeres con abortos durante los períodos preembriónico (< 6 semanas de gestación) y embriónico (6 a 9 semanas de gestación) tienen un SAF. Las pérdidas durante estas fases precoces de la gestación son comunes en la población general. Más características del SAF son las pérdidas fetales (>10 semanas de gestación) y los nacimientos prematuros, que se han relacionado con insuficiencia uteroplacentaria debida a trombosis localizada y quizás a una interferencia de los AAF con la anexina V (proteína anticoagulante placentaria) o con la propia invasión del trofoblasto.
Aproximadamente un 15% de las mujeres con abortos durante los períodos preembriónico (< 6 semanas de gestación) y embriónico (6 a 9 semanas de gestación) tienen un SAF. Las pérdidas durante estas fases precoces de la gestación son comunes en la población general. Más características del SAF son las pérdidas fetales (>10 semanas de gestación) y los nacimientos prematuros, que se han relacionado con insuficiencia uteroplacentaria debida a trombosis localizada y quizás a una interferencia de los AAF con la anexina V (proteína anticoagulante placentaria) o con la propia invasión del trofoblasto.
Otras manifestaciones clínicasSon también manifestaciones prominentes del SAF (~20%) la trombocitopenia, la anemia hemolítica y la livedo reticularis. Se utiliza el término de síndrome de Sneddon para designar la combinación de livedo reticularis e ictus, si bien los pacientes con este síndrome pueden no tener AAF. La presencia de livedo reticularis parece relacionarse también con un mayor riesgo de disfunción cognitiva en sujetos con SAF. La trombocitopenia no protege contra el desarrollo de eventos trombóticos.
Síndrome antifosfolipídico catastrófico
El término SAF “catastrófico” define una forma aguda y devastadora de SAF caracterizada por oclusiones vasculares múltiples y simultáneas que ocasionan un fracaso multiorgánico. Estos pacientes tienen en común: 1) evidencia clínica de afectación de múltiples órganos en un período inferior a una semana, 2) evidencia histopatológica de oclusión de pequeños vasos en al menos un órgano o tejido (una minoría tienen también trombosis de grandes vasos), y 3) confirmación de la presencia de AAF en el laboratorio.
Afortunadamente se trata de una variedad infrecuente de SAF (<1%), con una mortalidad, a pesar del tratamiento, del 50%. En más de la mitad de los casos se identifica un factor precipitante, por lo común una infección y, en menor medida, un procedimiento quirúrgico o el embarazo. La gran mayoría de estos pacientes se encuadran dentro de un SAFP. Los órganos más comúnmente afectados son por orden decreciente: riñón (hipertensión maligna), pulmón (distrés respiratorio, embolias pulmonares), cerebro (encefalopatía, infartos, convulsiones), piel (púrpura, livedo, necrosis cutánea), corazón (infarto, valvulopatía) y glándulas suprarrenales (insuficiencia suprarrenal aguda).
Diagnóstico
El término SAF “catastrófico” define una forma aguda y devastadora de SAF caracterizada por oclusiones vasculares múltiples y simultáneas que ocasionan un fracaso multiorgánico. Estos pacientes tienen en común: 1) evidencia clínica de afectación de múltiples órganos en un período inferior a una semana, 2) evidencia histopatológica de oclusión de pequeños vasos en al menos un órgano o tejido (una minoría tienen también trombosis de grandes vasos), y 3) confirmación de la presencia de AAF en el laboratorio.
Afortunadamente se trata de una variedad infrecuente de SAF (<1%), con una mortalidad, a pesar del tratamiento, del 50%. En más de la mitad de los casos se identifica un factor precipitante, por lo común una infección y, en menor medida, un procedimiento quirúrgico o el embarazo. La gran mayoría de estos pacientes se encuadran dentro de un SAFP. Los órganos más comúnmente afectados son por orden decreciente: riñón (hipertensión maligna), pulmón (distrés respiratorio, embolias pulmonares), cerebro (encefalopatía, infartos, convulsiones), piel (púrpura, livedo, necrosis cutánea), corazón (infarto, valvulopatía) y glándulas suprarrenales (insuficiencia suprarrenal aguda).
Diagnóstico
Como en otras enfermedades autoinmunes se han postulado una serie de criterios clínicos y de laboratorio que permiten clasificar a los pacientes con SAF. En la práctica se utilizan los reseñados en la Tabla 1. Sólo en aquellos pacientes con ACL IgG o IgM o AL negativos en los que se sospecha fuertemente un SAF se debería determinar la concentración de anticuerpos anti-beta-2-glucoproteína I o los ACL del isotipo IgA. Los AAF se pueden consumir durante un episodio trombótico agudo, por lo que es prudente medirlos 6 semanas después del mismo si resultaron inicialmente negativos. Asimismo, cualquier resultado positivo de AAF exige una confirmación al cabo de 3 meses para descartar que no se trate de un fenómeno transitorio, como el que se observa en asociación con determinadas infecciones virales.
Tabla 1: Criterios de clasificación del síndrome antiifosfolipídico
Tabla 1: Criterios de clasificación del síndrome antiifosfolipídico
| Criterios clínicos Trombosis vascular: • >1 episodios de trombosis arterial, venosa o de pequeño vaso Complicaciones obstétricas: • >1 muertes fetales después de 10 semanas de gestación • >1 nacimientos prematuros antes de 34 semanas • >3 abortos consecutivos no explicados antes de 10 semanas de gestación |
| Criterios de laboratorio • Anticuerpos anticardiolipina IgG o IgM en concentraciones superiores a 40 U, • Anticoagulante lúpico, • Anti-b2-glucoproteína I |
El diagnóstico de SAF exige al menos un criterio clínico y otro de laboratorio. La presencia de AAF se debe verificar en ³2 ocasiones con un intervalo de al menos 12 semanas
Diagnóstico diferencial
Se deben incluir en el diagnóstico diferencial del SAF otras enfermedades sistémicas (neoplasias, síndrome nefrótico, policitemia, trombocitosis), el uso de anticonceptivos orales o estados de trombofilia congénitos que predisponen al desarrollo de eventos trombóticos. El SAF, la homocisteinemia y la mutación 20210 del gen de la protrombina favorecen tanto las trombosis venosas como arteriales, mientras que el factor V de Leiden o las deficiencias de proteína C, proteína S o antitrombina III generalmente se manifiestan con trombosis venosas.
Diagnóstico diferencial
Se deben incluir en el diagnóstico diferencial del SAF otras enfermedades sistémicas (neoplasias, síndrome nefrótico, policitemia, trombocitosis), el uso de anticonceptivos orales o estados de trombofilia congénitos que predisponen al desarrollo de eventos trombóticos. El SAF, la homocisteinemia y la mutación 20210 del gen de la protrombina favorecen tanto las trombosis venosas como arteriales, mientras que el factor V de Leiden o las deficiencias de proteína C, proteína S o antitrombina III generalmente se manifiestan con trombosis venosas.
El diagnóstico diferencial de los abortos recurrentes también es amplio e incluye anomalías anatómicas, infecciones crónicas del tracto reproductor femenino, enfermedades sistémicas, desequilibrios hormonales (defectos de la fase lútea), anomalías cariotípicas maternas o paternas, anomalías genéticas fetales, abuso de substancias u otros estados procoagulantes.
Cuando existe un AL, el tiempo de protrombina es normal o sólo está ligeramente prolongado. Una prolongación manifiesta de este último puede deberse a un déficit de protrombina (factor II), una insuficiencia hepática o un déficit de vitamina K.
Tratamiento
Consideraremos el tratamiento del SAF en varios escenarios clínicos: sujeto asintomático con AAF, trombosis venosa o arterial instaurada, SAF catastrófico y pérdidas fetales recurrentes. Las pautas recomendadas en las tres primeras circunstancias se resumen en la Tabla 2.
Tromboprofilaxis primaria
Los individuos asintomáticos con AAF habitualmente se identifican al descubrirse un TTPA prolongado en un estudio de coagulación realizado por otros motivos (p.ej. un preoperatorio). Otro escenario frecuente es el paciente diagnosticado de LES, al que sistemáticamente se investiga la existencia de AAF, independientemente de que haya sufrido o no fenómenos trombóticos.
Los individuos asintomáticos con AAF habitualmente se identifican al descubrirse un TTPA prolongado en un estudio de coagulación realizado por otros motivos (p.ej. un preoperatorio). Otro escenario frecuente es el paciente diagnosticado de LES, al que sistemáticamente se investiga la existencia de AAF, independientemente de que haya sufrido o no fenómenos trombóticos.
En todo paciente con AAF es importante reducir o eliminar otros factores de riesgo para desarrollar trombosis como la estasis (inmovilización, cirugía), el uso de anticonceptivos orales o los factores tradicionales que favorecen la arteriosclerosis (tabaquismo, hipertensión, dislipemia, diabetes). La hidroxicloroquina podría proteger contra las trombosis en sujetos con LES y AAF. Aunque muchas de las personas con AAF y sin historia de trombosis pueden permanecer asintomáticas durante décadas, los expertos recomiendan administran 75 a 100 mg de aspirina diaria si se detecta AL o ACL a títulos altos de forma persistente.
Trombosis
El tratamiento inicial de una trombosis en sujetos con AAF no difiere del empleado en otras circunstancias (heparina y substitución posterior por anticoagulantes orales). Sin embargo, los pacientes con SAF tienen una alta probabilidad de recurrencia de la trombosis cuando se suspende la anticoagulación (50-70%). Por ello se sugiere prolongar el tiempo de anticoagulación más allá de los 3 a 6 meses estipulados. No obstante, no existe acuerdo sobre la duración exacta de la anticoagulación. Para algunos expertos debería indicarse de por vida tras un primer episodio de trombosis venosa o arterial. Otros, asumen la necesidad de profilaxis secundaria a largo plazo en pacientes que han sufrido una trombosis espontánea, una trombosis con compromiso vital (p.ej. embolia pulmonar, ictus, infarto de miocardio) o que poseen otros factores de riesgo vascular. Sin embargo, cuestionan la anticoagulación permanente si el primer episodio de trombosis venosa ha coincidido con una cirugía, con el uso de anticonceptivos orales u otra circunstancia protrombótica. Tampoco hay un consenso total sobre la intensidad de la anticoagulación. Probablemente son adecuados los dicumarínicos en régimen de intensidad moderada (INR: 2 a 3), aunque muchos expertos prefieren un régimen de intensidad elevada (INR>3). Al menos este último se debería indicar en pacientes con trombosis venosas recurrentes o con trombosis arteriales. No obstante, algunos estudios indican que tanto la warfarina en régimen de intensidad moderada (INR de 1,5 a 3) como la aspirina (325 mg/d) son opciones aceptables en pacientes con AAF y un primer episodio de ictus.
El tratamiento inicial de una trombosis en sujetos con AAF no difiere del empleado en otras circunstancias (heparina y substitución posterior por anticoagulantes orales). Sin embargo, los pacientes con SAF tienen una alta probabilidad de recurrencia de la trombosis cuando se suspende la anticoagulación (50-70%). Por ello se sugiere prolongar el tiempo de anticoagulación más allá de los 3 a 6 meses estipulados. No obstante, no existe acuerdo sobre la duración exacta de la anticoagulación. Para algunos expertos debería indicarse de por vida tras un primer episodio de trombosis venosa o arterial. Otros, asumen la necesidad de profilaxis secundaria a largo plazo en pacientes que han sufrido una trombosis espontánea, una trombosis con compromiso vital (p.ej. embolia pulmonar, ictus, infarto de miocardio) o que poseen otros factores de riesgo vascular. Sin embargo, cuestionan la anticoagulación permanente si el primer episodio de trombosis venosa ha coincidido con una cirugía, con el uso de anticonceptivos orales u otra circunstancia protrombótica. Tampoco hay un consenso total sobre la intensidad de la anticoagulación. Probablemente son adecuados los dicumarínicos en régimen de intensidad moderada (INR: 2 a 3), aunque muchos expertos prefieren un régimen de intensidad elevada (INR>3). Al menos este último se debería indicar en pacientes con trombosis venosas recurrentes o con trombosis arteriales. No obstante, algunos estudios indican que tanto la warfarina en régimen de intensidad moderada (INR de 1,5 a 3) como la aspirina (325 mg/d) son opciones aceptables en pacientes con AAF y un primer episodio de ictus.
Por último, advertir que la monitorización del nivel de anticoagulación en un pequeño grupo de pacientes con AL puede verse complicada por la interferencia que dicho autoanticuerpo tiene, en ocasiones, con el tiempo de protrombina y, en consecuencia, con el INR. Generalmente, este fenómeno se puede evitar seleccionando cuidadosamente el reactivo de tromboplastina empleado para la prueba del tiempo de protrombina.
La normalización de los AAF nunca es una indicación para suspender la anticoagulación, puesto que las concentraciones de estos autoanticuerpos varían ampliamente y los pacientes siguen teniendo riesgo trombótico independientemente de aquéllas.
Tabla 2: Recomendaciones terapéuticas en pacientes con AAF
Tabla 2: Recomendaciones terapéuticas en pacientes con AAF
| Situación cínica | Tratamiento |
| Asintomático | Evitar factores de riesgo vascular; no utilizar inhibidores COX-2; ningún tratamiento o dosis bajas de aspirina; hidroxicloroquina si LES sintomático. |
| Trombosis venosa o arterial | Anticoagulantes orales durante tiempo prolongado (indefinido); INR>3 en trombosis venosas recurrentes y arteriales1. |
| Trombosis recurrente a pesar de anticoagulación efectiva | Anticoagulantes orales (INR>3) + aspirina (100 mg/d). |
| SAF catastrófico | Tratar factores precipitantes (p.ej. antibióticos); heparina + dosis altas de corticoides + IGIV o plasmáféresis |
1Algunos estudios indican que la aspirina puede ser una alternativa a la anticoagulación oral en sujetos que sufren un primer episodio de isquemia cerebral. LES, lupus eritematoso sistémico; IGIV = inmunoglobulinas intravenosas
Tratamiento de otras manifestaciones clínicas
Los recuentos de plaquetas superiores a 50 x 109/μL generalmente no requieren terapia específica en ausencia de complicaciones hemorrágicas. Si aparece sangrado activo como consecuencia de una trombocitopenia intensa se indicarán corticoides (prednisona 1 mg/Kg/d). Si la situación es grave, se pueden administrar inicialmente pulsos intravenosos de metilprednisolona (1g/d durante 3 días). En casos refractarios, las IGIV (2g/Kg administrados en dosis divididas durante 2 a 5 días) pueden complementar la terapia corticoidea. Las trasfusiones de plaquetas en pacientes con SAF no son útiles, puesto que el mecanismo de la trombocitopenia es probablemente un daño plaquetar y se puede incrementar el riesgo de trombosis. Rara vez, la esplenectomía se deberá considerar en los casos refractarios a la corticoterapia.
Tratamiento de la embarazada con anticuerpos antifosfolipídicos
La mayor parte de recomendaciones sobre el tratamiento de la embarazada con AAF están basadas en la experiencia y opiniones de expertos más que en una sólida evidencia científica (Tabla 3). La evolución del embarazo en mujeres con AAF e historia de pérdidas gestacionales previas puede mejorar significativamente con la combinación de dosis bajas de aspirina (75-100 mg/d) y heparina (las de bajo peso molecular –HBPM- son las más utilizadas). El tratamiento se inicia tan pronto como se diagnostique el embarazo (βHCG +), aunque algunos abogan por su prescripción antes de la concepción. Aquellas pacientes que estén tomando anticoagulantes orales por una trombosis previa y quieran quedar embarazadas deberán sustituir lo más precozmente posible el acenocumarol o la warfarina por heparina subcutánea, ya que los primeros son teratogénicos si el feto queda expuesto en las primeras 6-12 semanas de gestación. Toda embarazada que reciba heparina debe prevenir la osteoporosis con la suplementación adecuada de calcio y vitamina D.
La monitorización ecográfica del crecimiento fetal y del flujo sanguíneo uteroplacentario durante la gestación es crucial porque ayuda a decidir el momento del parto. No se conocen anomalías genéticas fetales asociadas a AAF y la probabilidad de que el niño tenga este tipo de autoanticuerpos es baja.
Las IGIV mensuales (2 g/Kg) son un posible tratamiento adicional para las mujeres en las que ha fracasado previamente la combinación de aspirina y heparina.
Tabla 3: Tratamiento de las pacientes embarazadas con AAF
Tratamiento de otras manifestaciones clínicas
Los recuentos de plaquetas superiores a 50 x 109/μL generalmente no requieren terapia específica en ausencia de complicaciones hemorrágicas. Si aparece sangrado activo como consecuencia de una trombocitopenia intensa se indicarán corticoides (prednisona 1 mg/Kg/d). Si la situación es grave, se pueden administrar inicialmente pulsos intravenosos de metilprednisolona (1g/d durante 3 días). En casos refractarios, las IGIV (2g/Kg administrados en dosis divididas durante 2 a 5 días) pueden complementar la terapia corticoidea. Las trasfusiones de plaquetas en pacientes con SAF no son útiles, puesto que el mecanismo de la trombocitopenia es probablemente un daño plaquetar y se puede incrementar el riesgo de trombosis. Rara vez, la esplenectomía se deberá considerar en los casos refractarios a la corticoterapia.
Tratamiento de la embarazada con anticuerpos antifosfolipídicos
La mayor parte de recomendaciones sobre el tratamiento de la embarazada con AAF están basadas en la experiencia y opiniones de expertos más que en una sólida evidencia científica (Tabla 3). La evolución del embarazo en mujeres con AAF e historia de pérdidas gestacionales previas puede mejorar significativamente con la combinación de dosis bajas de aspirina (75-100 mg/d) y heparina (las de bajo peso molecular –HBPM- son las más utilizadas). El tratamiento se inicia tan pronto como se diagnostique el embarazo (βHCG +), aunque algunos abogan por su prescripción antes de la concepción. Aquellas pacientes que estén tomando anticoagulantes orales por una trombosis previa y quieran quedar embarazadas deberán sustituir lo más precozmente posible el acenocumarol o la warfarina por heparina subcutánea, ya que los primeros son teratogénicos si el feto queda expuesto en las primeras 6-12 semanas de gestación. Toda embarazada que reciba heparina debe prevenir la osteoporosis con la suplementación adecuada de calcio y vitamina D.
La monitorización ecográfica del crecimiento fetal y del flujo sanguíneo uteroplacentario durante la gestación es crucial porque ayuda a decidir el momento del parto. No se conocen anomalías genéticas fetales asociadas a AAF y la probabilidad de que el niño tenga este tipo de autoanticuerpos es baja.
Las IGIV mensuales (2 g/Kg) son un posible tratamiento adicional para las mujeres en las que ha fracasado previamente la combinación de aspirina y heparina.
Tabla 3: Tratamiento de las pacientes embarazadas con AAF
| Escenario clínico | Recomendación terapéutica |
| AAF, sin historia de SAF (no pérdidas gestacionales ni trombosis) | Ningún tratamiento o dosis bajas de aspirina; suspender aspirina 3-5 días antes del parto |
| Historia de abortos recurrentes precoces, pérdidas fetales, neonatales o preeclampsia severa (sin historia de trombosis vasculares) | Dosis bajas de aspirina + HBPM dosis profilácticas (p.ej enoxaparina 1mg/Kg/d)1; suspender aspirina 3-5 días y heparina 6-24 horas antes del parto; reanudar heparina 6-8 horas después del parto y mantener 6 semanas |
| Historia de abortos recurrentes precoces, pérdidas fetales, neonatales o preeclampsia severa (con historia de trombosis vasculares) | Dosis bajas de aspirina + HBPM dosis terapéuticas (p.ej. enoxaparina 1mg/Kg/12h)2; suspender aspirina 3-5 días y heparina 6-24 horas antes del parto; reanudar de nuevo anticoagulantes orales (o heparina) 6-8 horas después del parto |
1Algunos expertos recomiendan sólo aspirina si la historia es de pérdidas precoces y reservan la combinación para los casos en que ésta fracasa. Asimismo, otros utilizan dosis mayores de enoxaparina (1 mg/Kg/12 h), especialmente si las pérdidas son tardías.
2Si la paciente estaba anticoagulada se debe sustituir la anticoagulación oral por HBPM antes de la concepción o en cuanto se diagnostique el embarazo.
AAF, anticuerpos antifosfolipídicos; SAF, síndrome antifosfolipídico; HBPM, heparina de bajo peso molecular
2Si la paciente estaba anticoagulada se debe sustituir la anticoagulación oral por HBPM antes de la concepción o en cuanto se diagnostique el embarazo.
AAF, anticuerpos antifosfolipídicos; SAF, síndrome antifosfolipídico; HBPM, heparina de bajo peso molecular
Puntos Claves
• Unos anticuerpos anticardiolipina positivos pueden ser un fenómeno transitorio relacionado con una infección aguda u otro proceso.
• Unos anticuerpos anticardiolipina positivos pueden ser un fenómeno transitorio relacionado con una infección aguda u otro proceso.
• Se debe considerar la posibilidad de un SAF en las siguientes circunstancias: trombosis venosa o arterial inexplicada, trombosis en lugares inusuales, trombosis en personas jóvenes, trombosis recurrentes o mujer con pérdidas fetales o preeclampsia.
• Se deben utilizar al menos dos pruebas para detectar AAF: el AL y los ACL, ya que los pacientes pueden tener negativa una de ellas y positiva la otra.
• La terapia anticoagulante puede interferir con la detección de AL.
• La terapia anticoagulante puede interferir con la detección de AL.
• Para descartar fehacientemente la existencia de un AL, al menos dos pruebas de la coagulación dependientes de fosfolípidos (p.ej. TTPA y TVVR) deben ser negativas.
• La prolongación del TTPA o el tiempo de protrombina ligeramente prolongado como consecuencia de AAF no supone un riesgo aumentado de sangrado y, por tanto, no son una contraindicación para llevar a cabo procedimientos quirúrgicos.
• Las trombosis en pacientes con AAF tienen una probabilidad elevada de recurrir, por lo que se recomienda anticoagulación prolongada.
• Ni la trombocitopenia ni el TTPA prolongado protegen contra las trombosis.
• La progresión de SAFP hacia LES es infrecuente.
• En mujeres con LES, AAF e historia de abortos recurrentes, un nuevo embarazo se debe abordar con la combinación de aspirina y HBPM.
• La prolongación del TTPA o el tiempo de protrombina ligeramente prolongado como consecuencia de AAF no supone un riesgo aumentado de sangrado y, por tanto, no son una contraindicación para llevar a cabo procedimientos quirúrgicos.
• Las trombosis en pacientes con AAF tienen una probabilidad elevada de recurrir, por lo que se recomienda anticoagulación prolongada.
• Ni la trombocitopenia ni el TTPA prolongado protegen contra las trombosis.
• La progresión de SAFP hacia LES es infrecuente.
• En mujeres con LES, AAF e historia de abortos recurrentes, un nuevo embarazo se debe abordar con la combinación de aspirina y HBPM.
 El examen debe tener en cuenta los antecedentes personales. En las manos buscar cambios de color, cambios en el lecho ungular y en la integridad de la piel. La esclerodactilia, las deformidades en flexión, las fricciones tendinosas y la calcinosis se ven en la esclerosis sistémica. Las úlceras digitales no son normales y siempre reflejan el fenómeno de Raynaud secundario; estas úlceras deben motivar enseguida el examen de otros signos de una enfermedad del tejido conectivo y la derivación al especialista.
El examen debe tener en cuenta los antecedentes personales. En las manos buscar cambios de color, cambios en el lecho ungular y en la integridad de la piel. La esclerodactilia, las deformidades en flexión, las fricciones tendinosas y la calcinosis se ven en la esclerosis sistémica. Las úlceras digitales no son normales y siempre reflejan el fenómeno de Raynaud secundario; estas úlceras deben motivar enseguida el examen de otros signos de una enfermedad del tejido conectivo y la derivación al especialista.